Molecular Interaction Surfaces for Hydrogen Binding
Category
Modeling/Simulation
Laboratory
Lawrence Berkeley National Laboratory (LBNL)
Description
Within the computational framework of HyMARC, we are developing molecular interaction surfaces that resolve the nature of molecular hydrogen’s interactions with nanoporous materials. Hydrogen can either be physisorbed or chemisorbed within the vast internal surface area provided by MOFs and COFs. For high-density storage, a regime that is intermediate between strong chemisorption and weak physisorption is sought. This calls for optimization of local on-site interactions and resolution of stabilizing influences in chemically meaningful terms.
Energy decomposition analysis (EDA) offers a chemically intuitive unraveling of hydrogen gas interactions with the local environment around its binding site in a nanoporous framework. Molecular hydrogen may interact with the framework electrostatically, via instantaneous dipole-induced dipole or van der Waal’s interactions, and through polarization and charge transfer. EDA tools provide a quantitative delineation of such interactions and a precise account of interacting molecular orbitals at play in hydrogen storage. The charge transfer term in EDA analysis provides a definitive estimate for the proclivity of a material to chemisorb hydrogen. This makes such decomposition schemes key to deciphering the nature of the interactions and offer signatures that can then be harnessed to design the best material for storage applications. In combination with Q-Chem’s state-of-the-art range-separated hybrid DFT functionals, this feature is a potent computational recipe for identifying new families of storage materials.
Diverse molecular interactions can be quantified with energy decomposition analysis with absolutely localized molecular orbitals. However, such analyses are performed at a single geometry and decompose a “single point” interaction energy. As a consequence, physically meaningful EDA components on the molecular electronic structure are not directly obtained. Potential energy surfaces (PESs) have been optimized at the frozen, polarized, and fully relaxed levels of density functional theory to address this gap. The ensuing ALMO-EDA in the adiabatic picture has energy contributions that are drawn from energy differences between the stationary points on between these potential energy surfaces. What ensues is a chemical intuitive decomposition of molecular interaction surfaces, with each surface having characteristic vibrational frequencies. This brings stabilization effects out of electronic energy into the more measurable form of molecular vibrations. We are working towards bringing insight from these chemical handle-based molecular interactions into the study of gas sorption in nanoporous materials.
Status
Online and available for use in collaboration with HyMARC.
Figures
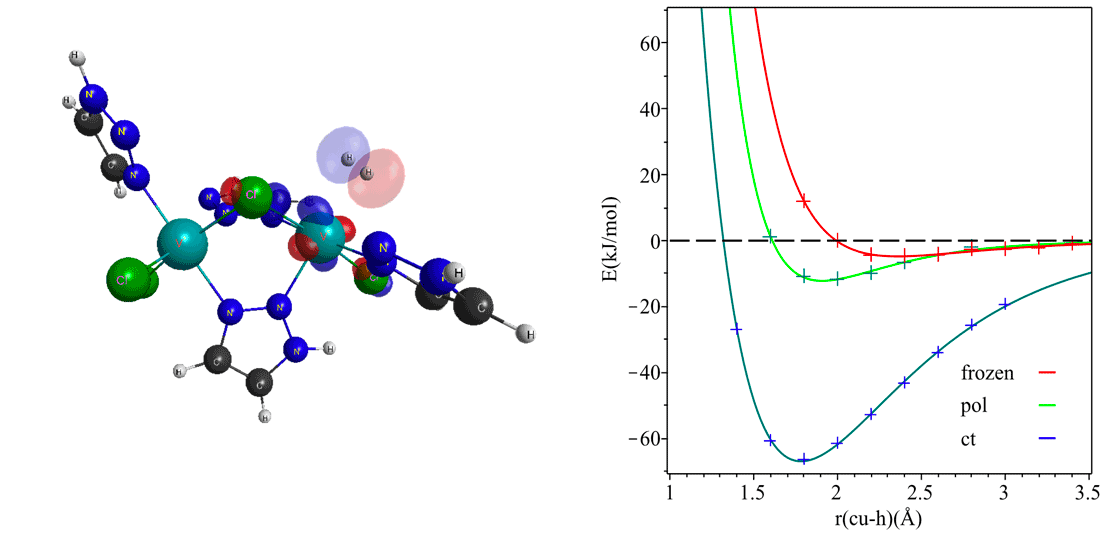
Figure 1. a) Significant charge transfer orbitals for back-donating interactions between molecular hydrogen and V(II). b) Molecular interaction surface of hydrogen interacting with a bare Cu(I) site.
References
- Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A 111 (2007): 8753.
- Mao, P. R. Horn and M. Head-Gordon, Phys. Chem. Chem. Phys. 19 (2017): 5944.